
.jpg "新聞中心")

反應(yīng)操作、柱層析、蒸餾等實(shí)驗(yàn)室工作經(jīng)驗(yàn)分享
發(fā)布時間:2023-05-19瀏覽次數(shù):1843
實(shí)驗(yàn)室工作有很多是經(jīng)驗(yàn)性的,有很多細(xì)節(jié)是課本上沒有的,但是在實(shí)驗(yàn)室實(shí)際應(yīng)用中又非常重要,本文我總結(jié)了一些自己在多年的實(shí)驗(yàn)室工作經(jīng)驗(yàn),主要是一些個人習(xí)慣。良好的實(shí)驗(yàn)室習(xí)慣的養(yǎng)成,往往會是一個很好的開始,它是避免人為反應(yīng)失敗的[敏感詞]的利器,也是提高效率,避免人力浪費(fèi)的很好的助力。
[敏感詞]部分:反應(yīng)操作
1,原則上所有的反應(yīng)都不能用塞子塞住,從而造成反應(yīng)在狹小的密閉空間下反應(yīng),必須在出口處街上氮?dú)馇蚧蛘呷ǎㄌ厥獾娜绺邏悍磻?yīng)或 sealed tube 反應(yīng)除外),以免因反應(yīng)放熱或產(chǎn)生氣體而發(fā)生沖料或者發(fā)生爆炸。
2,處理反應(yīng)時選擇合適的容器,[敏感詞]在預(yù)估的所有體積的二到三倍之間,以免出現(xiàn)容器過滿的狀況。
3,任何反應(yīng)若非會放出含 H+之物質(zhì),均應(yīng)盡可能在惰氣(氮?dú)饣驓鍤猓┫到y(tǒng)下操作,以避免不必要之副反應(yīng)(side reaction)。如必須于惰氣系統(tǒng)下操作則以使用惰氣氣球系統(tǒng)為宜。
4,標(biāo)準(zhǔn)惰氣系統(tǒng)反應(yīng)處理方式是將反應(yīng)器皿于烘箱中取出后迅速放入攪拌子并連接氮(氬)氣出口之后迅速將反應(yīng)所需設(shè)備組合起來,并用火焰干燥,然后抽真空再灌入氮(氬)氣,并重復(fù)兩次。
5,任何一未知反應(yīng),除非有極近似之反應(yīng)作參考,否則應(yīng)從0℃或室溫開始嘗試,若不反應(yīng)再逐步升高其溫度,反之若反應(yīng)太快有副作用時,再降低其溫度。
6,任何一未知反應(yīng)于 set up 完成后30 分鐘內(nèi)即應(yīng)檢查反應(yīng)進(jìn)行之情況,我們可取適量之原液(Aliquots)以TLC,NMR,GC,IR 或其他適當(dāng)?shù)募记蓹z驗(yàn)之。確定其反應(yīng)進(jìn)度后,才可決定是否在原條件下反應(yīng),切不可任意由其反應(yīng),或work-up 而不予檢查。
7,檢查反應(yīng)時若發(fā)現(xiàn)超過 3 小時沒有任何變化(反應(yīng))發(fā)生則可嘗試逐漸升高溫度10-30℃,然后再等十分鐘后檢查其變化,若再過一小時后仍無反應(yīng)則再升高溫度10-30℃,如此反復(fù)嘗試直到反應(yīng)發(fā)生為止。
8,任何一反應(yīng)若需加熱或反應(yīng)中可能生熱時必須加裝冷凝管以確保物質(zhì)不會揮發(fā)散失。
9,若反應(yīng)之溶劑為低沸點(diǎn)物質(zhì)(如乙醚,二氯甲烷等)則冷凝管內(nèi)需通冰水,否則溶劑可能揮發(fā)散失,尤其在加熱回流時,不注意會造成反應(yīng)物濃度過高,甚至干涸。
10,怕水而需低溫反應(yīng),先Set-up 好所有的equipment、solvent,及部分不會產(chǎn)生反應(yīng)之試劑,最后才以冷媒冷卻反應(yīng)系統(tǒng),否則在set-up 時易有水汽進(jìn)入,影響反應(yīng)效果。
11,如果反應(yīng)之產(chǎn)物 NMR 無法解釋,應(yīng)立即點(diǎn)TLC 以檢查其純度,如果不純則NMR圖譜之積分自然無法解釋,應(yīng)純化后再測。
12,任何新反應(yīng)在完全定案前,所有嘗試反應(yīng)之產(chǎn)物,請務(wù)必予以保留,即使非我們所預(yù)期的產(chǎn)物或混合物,均有保留及比較TLC 的價值。
13,反應(yīng)后務(wù)必將所有“有意義”之TLC plates 按原尺寸畫于筆記薄上,以便以后比較用。
14,下班前應(yīng)盡力完成第二天實(shí)驗(yàn)之準(zhǔn)備工作,如明日需用之干燥玻璃器皿,設(shè)備,或找好需用的試劑及溶劑等。
15,如預(yù)期反應(yīng)需超過 12 小時,宜盡量利用下午或晚上set up 反應(yīng)。如預(yù)期反應(yīng)需超過36 小時,宜盡量利用周5 或周6 set up,以求節(jié)省時間提高效率。
第二部分:柱層析
1,一般而言極性大的物質(zhì)在極性固定相上的移動速率最?。ㄒ蚱湮敏感詞])反之極性小的物質(zhì)在極性固定相上的移動速率[敏感詞]。但所有物質(zhì)使用高極性流動相時在極性靜相時之移動速率均增大。故依各分離物質(zhì)的極性選擇極性適當(dāng)?shù)墓潭ㄏ嗉傲鲃酉嗌踔疗溆昧烤鶠閷游觯–hromatography)成功之關(guān)鍵。

2,任何化學(xué)物若以 TLC 鑒定其純度,其所顯示的Rf 值超過0.7 或低于0.1 時,均無法確定其是否為單一點(diǎn)(one spot),因?yàn)樵诖朔NRf 值時,許多Rf 較接近的點(diǎn)會重合。因此我們所選擇觀察物最適合之Rf 值為0.3-0.5。
3,Run Column 時所選用之solvent 系統(tǒng)應(yīng)配合選用硅膠(stationary phase)的用量及sample(樣品)的量來選擇其極性組合,一般而言欲分離之主樣品Rf 值以0.15-0.25為宜。
4,一般最常用之 Solvent 依其極性大小依次為n-Hexane(or pet.ether)<EtOAc<MeOH,組合為最通用,其他如Ether 也常為中極性之溶劑,對于氨類氯化溶劑如CH2Cl2,CHCl3常有特殊的效果,而Benzene 對含芳香性之化合物也會有出人意外之好效果。
5,Run Column 所用之Solvent 在配好后,應(yīng)以TLC 再作檢測一次,以確定是否為恰當(dāng)之Solvent 系統(tǒng)。尤其若需要收集一點(diǎn)時,則在分離前之混合物必須留一些以便作比較參考用。
6,在填裝層析管(packing column)時若不慎有氣泡(如Solvent 部分流干)可補(bǔ)滿Solvent后以木棒(或簽字筆)輕敲管壁,讓氣泡浮出。因?yàn)闅馀葜袥]有硅膠,分離物流經(jīng)此處時移動速度會比其他處快,而造成此區(qū)域分離物與其他無氣泡處之分離物排列次序混亂之現(xiàn)象,影響分離效果。
7,在 Run Column 時所收集的分離份(fraction)體積(毫升)通常為硅膠克數(shù)的1/4 以下。實(shí)際情況可用下列公式來求出約略值,如欲分離成分A 與B,而A 之Rf 值為0.3,B 之Rf 值為0.2 則最適合之分離份(fraction)體積為:設(shè)硅膠量為50g
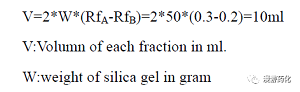
8,在作層析時,如何濃密而又均勻的將樣品 Loading 在起始點(diǎn)乃是層析分離成功與否的關(guān)鍵。通常的做法是將樣品溶解在不超過其體積兩倍之低極性溶液,然后小心的將樣品涂布于起始點(diǎn)。柱層析(Column Chromatography)時務(wù)必等樣品液完全進(jìn)入固定相(Stationary phase)后,以少許溶劑再將管壁上沾著的樣品沖入固定相后,才可正式加溶劑開始 Run Column。由于大部分化合物在低極性溶劑中溶解度很差,我們也可以將樣品均勻地與少量硅膠混合后干法上樣。這也是我們采用最多的方法。
9,在 Run Column 時切記不可使Column 內(nèi)溶劑過少而使靜相頂端干涸,且在添加溶劑也要小心,不可使溶劑急沖而下破壞靜相之頂層,尤其在剛開始Run Column 時,否則將造成樣品之過量稀釋分散,從而嚴(yán)重影響層析效果。
10,在收集時,可以先行以 Rf 值以固定相之總量計(jì)算大約樣品出現(xiàn)時之流出溶劑體積,在此體積達(dá)1/2 量前可以大瓶收集,1/2 量之后再以預(yù)定之小容器分別收集。
11,Run Column 時Solvent 之流速一般而言(HPLC,MPLC 除外),流速均以越快越好,因?yàn)橐话愣訫obile Phase Velocity 與Plate height 成反比,而Plate heigh 越小越好,故動相流速是越快越好。故Flash Chromatography 之效果常較一般Gravity column 為佳。
第三部分:蒸餾
1,蒸餾時務(wù)必先判斷你們樣品在其沸點(diǎn)時是否會分解,否則就必須選擇減壓蒸餾或其他的分離方法。
2,一般實(shí)驗(yàn)室的蒸餾僅用于分離沸點(diǎn)差 50℃以上物質(zhì)。(通常只是用于除去溶劑或有色的高分子粘滯物)。如欲分離沸點(diǎn)較接近之物質(zhì)則需加裝分餾管,而分餾管之長度即裝填物需視分離之物質(zhì)沸點(diǎn)接近之程度而定。一般而言,沸點(diǎn)差小于20℃或總量小于1 克,則需特殊裝置(如Spinning band or molecular distillation)否則便無法做分餾了。
3,蒸餾時其外加熱媒之溫度與蒸出沸點(diǎn)差至少會在 30℃左右。
4,在做減壓蒸餾前,務(wù)請查明所有欲蒸餾物的沸點(diǎn),以免于壓力速降時造成‘突沸’而造成整個樣品沖出。尤其需注意溶劑是否已在Rotary evaporator 上抽干。
5,如果無參考圖表,可約略以下法計(jì)算各物質(zhì)之低壓沸點(diǎn)。通常壓力由760mmHg 減至25mmHg 其沸點(diǎn)降低100℃,其后壓力每降低一半,其沸點(diǎn)降低10℃。
6,若兩個或更多物質(zhì)混合時,通常會形成各種成分之共沸物。而在較低的溫度即沸騰汽化而出。此種情形最常發(fā)生于樣品含溶劑的狀況。
7,減壓蒸餾時,務(wù)必加裝冷卻 Trap,否則不但可能因樣品被吸入泵而失去樣品,也會損壞真空泵。
第四部分:其他日常操作
1,使用樣品后盡可能將其歸還回原位,尤其需冷藏或干燥者必須封好后放回原位。所有裝于燒瓶內(nèi)之化合物,應(yīng)隨時標(biāo)明編號(以膠帶粘貼以便清除)。
2,烘箱非儲藏柜僅僅用于烘干玻璃器皿,烘干后需在近期內(nèi)(一天內(nèi))使用者才放入,其余物品均應(yīng)放于抽屜或涼干架。
3,如果 Solvent 或低沸點(diǎn)物(bp<150℃)能在Rotary evaporator 上清除者,必須先在Rotaryevaporator 上處理至不能再濃縮后,才可放上Vacuum pump。否則低沸點(diǎn)物可能被抽入泵中。
4,使用 Vacuum pump 必須要干冰作Trap 之冷媒,使用前必檢查Trap 內(nèi)是否有殘留物,若有,則需清除后方可使用。并在使用完后立即清理Trap 之廢物。
5,萃取時選用之 Solvent 應(yīng)注意其比重,若其比重大于1(如CH2Cl2, CHCl3, CCl4)而反應(yīng)原液為比重小于1 之溶劑,(如Ether, THF 或Benzene)時,切不可同時混入分液漏斗,否則會有乳化不分層之現(xiàn)象,造成分離困難。
6,當(dāng)溶液裝于有磨口的容器內(nèi)時,在任何操作狀況時,均應(yīng)避免使溶液接觸到磨口,因?yàn)楫?dāng)溶劑揮發(fā)后溶質(zhì)將殘留在磨口上,會使你損失所要的Compound,而在加蓋常會有打不開之現(xiàn)象。所以若不慎或有不得以有溶液接觸磨口,則應(yīng)以純?nèi)軇┙櫧佑|部分,務(wù)使溶劑殘留之痕跡消除為止。
7,實(shí)驗(yàn)記錄本應(yīng)包含反應(yīng)方程式,參考文獻(xiàn),試藥用量(Weight,Mole),理論產(chǎn)率,實(shí)驗(yàn)步驟,TLC,光譜記錄,與注意事項(xiàng)或結(jié)論。
8,實(shí)驗(yàn)室對量的記載應(yīng)盡量保持三為數(shù)字,以求其[敏感詞]性;對不滿 1 之單位應(yīng)用下一級之單位。如0.1 克應(yīng)寫為100mg,0.1mol 應(yīng)寫為100mmol。
9,實(shí)驗(yàn)本務(wù)必按進(jìn)度如實(shí)記錄,要如實(shí)做到‘做到哪里,寫到哪里’,而且不得帶出實(shí)驗(yàn)室。實(shí)驗(yàn)記錄務(wù)必跟隨實(shí)驗(yàn)進(jìn)度將所有事實(shí)詳細(xì)記錄,成功之反應(yīng)必有產(chǎn)物,產(chǎn)率,顏色,狀態(tài)及純化方式,失敗之實(shí)驗(yàn)必說明鑒定失敗的方法及證據(jù),并盡可能說明原因。